◤会员文:顾名思医◢脊髓性肌肉萎缩症新药 罕见病患迎新曙光



脊髓性肌肉萎缩症只能等死?我国核准新药上市,让罕见病看见新曙光!
ADVERTISEMENT
ADVERTISEMENT

呼吸、吞咽、行走、坐立…这些一般人与生俱来的本能,却是某些孩子(甚至成人)求之不得的奢望。他们坐在轮椅、躺在病床,有些甚至需要仰赖各种设备才能呼吸或继续生存,即使日复一日努力复建,也未必能恢复基本生活功能,只能清醒地看着生命功能一点一滴流逝。这群人的症状和严重程度未必相同,但他们都有一个共同的名字——脊髓性肌肉萎缩症(SMA)患者。

这种病会导致患者肌肉无力及逐渐丧失运动能力,专门研究临床遗传学的小儿专科医生庄玉秀直言,目前疾病治疗方面仍存在重大的未满足医疗需求,尤其是与这种情况共存的成年人:“患有脊髓性肌肉萎缩症的儿童,可能在出生后前几天就开始出现症状,而且会随着时间推移,逐渐失去肌肉力量,因此在肌肉开始变弱之前,接受早期治疗至关重要。虽然发病年龄、症状范围和严重程度因人而异,但脊髓性肌肉萎缩症所带来的生理和情感挑战,会对整个家庭造成影响。过去没有医学进展能缓解脊髓性肌肉萎缩症的衰弱症状,直到最近才有了更多的治疗选项。”
庄医生分析,虽然我国核准了脊髓性肌肉萎缩症的新药治疗,但患者仍存在许多未获满足的需求:“这种疾病会对患者和他们的家庭成员造成各种影响,除了疾病本身严重影响日常生活,他们也需要面对心理健康(例如:压力、焦虑、照顾者的无力感)、经济财务等各方面问题。脊髓性肌肉萎缩症的治疗和管理方案并不只是药物,更包括多学科护理和协作,患者和医护人员通常需要关注的领域包括呼吸系统护理、康复治疗、职业治疗、营养、骨科、精神健康、缓解治疗等等。”
另外,她也认为应该通过各种教育宣导,提高人们对于脊髓性肌肉萎缩症的认识;以及通过产前筛查和推动SMNI突变基因携带者筛查或新生儿筛查,及早诊断脊髓性肌肉萎缩症。
有时候,患者未必害怕死亡,他们更担心成为一个无康复希望的患者,无法独立生活,只能拖累家人的存在。因此,在应对脊髓性肌肉萎缩症这个课题上,除了医学和药物研究需要持续进步,以及政策方面的制定,也需要社会人士给予罕见病群体更多的关爱和理解,让患者的生命之路能够越走越宽。

认识脊髓性肌肉萎缩症
你知道吗?根据估计,每6000至一万新生婴儿中,就有 1人受到脊髓性肌肉萎缩症(Spinal Muscular Atrophy,简称SMA)影响,而且这种罕见病,也是导致全球婴儿死亡的首要遗传性原因之一。5q型脊髓性肌肉萎缩症是最常见的疾病类型,大约占所有SMA病例的95%。
脊髓性肌萎缩症是一种可能致命、严重渐进性的神经肌肉疾病。脊髓性肌萎缩症是由运动神经元存活基因1(简称SMN1) 突变所造成,这种基因主要负责制造运送神经元存活(SMN) 蛋白,这种蛋白质遍布全身,且对控制肌肉和维持运动神经功能至关重要。基因突变会导致SMN蛋白缺乏,缺少了这种蛋白,神经细胞就无法正常运作,并逐渐衰退,随着时间推移,就会导致肌肉无力。
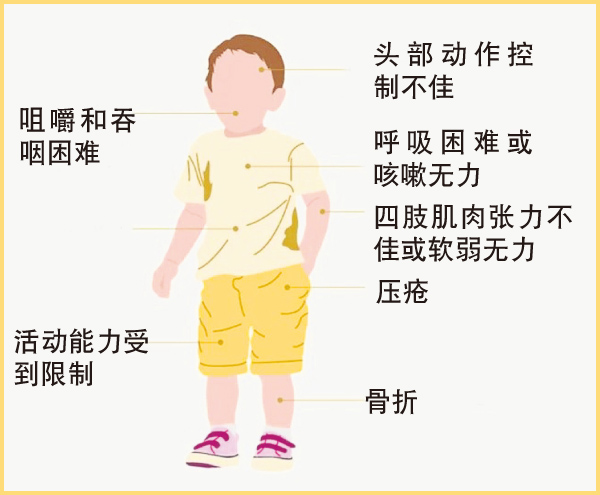
脊髓性肌肉萎缩症对每个患者造成的影响都不同,严重程度通常与发病年龄相关。目前,临床上主要根据不同发病年龄及所能达到的最高运动功能(例如:独自坐稳、站立或行走),将脊髓性肌肉萎缩症分为4大类型:I型、II型、III型和IV型。无论如何,这些“类型”并非严格的类别区分,因为即使是同一类型患者,其病发速度及症状也可能有很大的差异。根据不同的脊髓性肌肉萎缩症类型,患者的个人体力,以及行走、进食或呼吸能力都可能会显著减弱或丧失。脊髓性肌肉萎缩症的影响,主要集中在近端和躯干肌肉,但也可能会影响膈肌和远端肌肉的四肢,但这种病不会影响患者的智力发展。发病年龄从出生到成年皆有可能。
孕前宜做基因检测
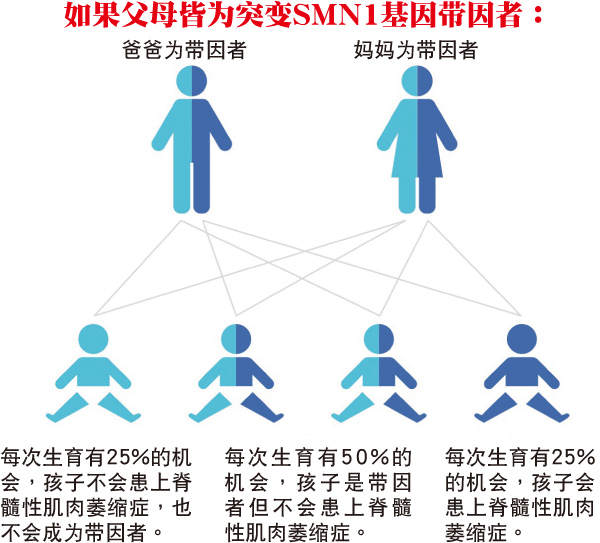
脊髓性肌肉萎缩症为一种常染色体(指染色体组中除性染色体以外的染色体)隐性疾病,意思是患者必需同时遗传到来自父母的突变SMN1基因,才有可能罹患脊髓性肌肉萎缩症。如果孩子只遗传到一组突变的SMN1基因,他们将会被视为“带因者”,但通常没有脊髓性肌肉萎缩症的症状。
SMA基因病变携带者一般没有症状,但有缺陷的基因可能会遗传给他们的孩子。所以,如果一个人有脊髓性肌肉萎缩症的家族病史,其成为带因者的几率将高于平均值。建议孕前或决定生育时咨询医生有关遗传性疾病的相关事项,或在怀孕初期即安排检查并做后续必要的遗传咨询。
脊髓性肌肉萎缩症需要综合多方面资讯才能诊断,其中包括专业医生评估、肌电图和神经传导检查、血液检测(用于检查异常基因),甚至必要时需做肌肉活检等。无论如何,当婴幼儿出现无法解释的肌无力和肌肉萎缩情况时,医生通常会进一步检查患有脊髓性肌肉萎缩症的可能性。
脊髓性肌肉萎缩症患者的症状未必相同,且会依据发病年龄和功能状态分成不同类型。即使是同一种类型,也存在着不同的严重程度,部分患者也可能没有明确类型。以下是一些患者可能出现的症状:

Deepti Saraf:冀新药改善治疗效果
马来西亚国家药品管理局(简称NPRA)日前批准将EvrysdiTM(risdiplam)应用于治疗年龄2个月及以上的脊髓性肌肉萎缩症患者。庄玉秀医生指出,药物的研发让脊髓性肌肉萎缩症患者及其家人看到了更多希望曙光,同时能够更好地帮助他们克服伴随疾病而来的各种挑战。
研发上述药物的罗氏集团(Roche)马来西亚分公司总经理Deepti Saraf认为,新药将为这种罕见神经系统疾病的患者带来意义重大的效益:“我们期待与医疗保健领域携手合作,提高人们对这种疾病的意识,并希望改善治疗结果。另外,我们也恳切呼吁政策制定者和保险供应商确保制定明确及具承诺性的政策方向,其中包括法规和奖励措施,以及制定反基因歧视的保险条款。”她希望未来不同单位都能携手合作,提高社区教育和醒觉意识,同时也推动脊髓性肌肉萎缩症疾病的更多相关研究。
risdiplam是一种运动神经元存活2(简称SMN2)剪接修饰剂,旨在治疗由染色体5q突变导致SMN 蛋白缺乏而引起的脊髓性肌肉萎缩症。这种液态型药物可以在家中每天通过口服或经由胃食管给药。此药物的核准基于一项涵盖与脊髓性肌肉萎缩症共存的广泛疾病人群临床研究结果–SUNFISH。研究对象涵盖了年龄2至25岁、有症状II型和III型儿童和成人患者。SUNFISH是首个及唯一一个纳入II型和III型脊髓性肌肉萎缩症成人患者的安慰剂对照研究。

ADVERTISEMENT
ADVERTISEMENT